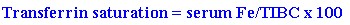
This is a disease of excess iron storage with resultant deposition of iron in the lysosomes of parenchymal cells of various visceral organs. There are no excretory mechanisms for excess iron stores. The normal metabolic balance between iron in the body and its environment is maintained by controlling the amount of iron that is absorbed in the intestine. Patients with hemochromatosis have lost this regulatory mechanism, and therefore accumulate excessive amounts of total body iron stores up to five times the normal value. Usual organs affected include the liver, pancreas, heart, joints and endocrine organs (to include the adrenal, thyroid, parathyroid, and pituitary glands). Clinical manifestations include weakness, lassitude, weight loss, change in skin color, abdominal pain, loss of libido, impotence, amenorrhea, arthropathy, and symptoms related to the onset of diabetes mellitus. These symptoms are the most frequently encountered initial symptoms although the disease is classically associated with liver disease (this is often the first manifestation), diabetes mellitus, and hyperpigmentation. More established disease states are characterized by hepatomegaly, hyperpigmentation, spider angiomata, splenomegaly, arthropathy, ascites, cardiac arrhythmias, congestive heart failure (CHF), loss of body hair, testicular atrophy, and jaundice. Excessive parenchymal iron acts as a carcinogen, and patients often manifest hepatomas as the most frequent cause of death followed by CHF, hepatic coma, pneumonia, and hematemesis secondary to variceal bleeding. Infection is a common and potentially life-threatening complication, and is often secondary to Vibrio vulnificus (commonly found in coastal water shellfish) and Yersinia enterocolitica, which may induce spontaneous peritonitis. Secondary iron overload results from repeated blood transfusions, alcoholic related disease, and porphyria cutanea tarda.
Hemochromatosis is a genetic disease with transmission being recessive, and homozygosity is necessary for clinical expression. The frequency of the disease is approximately 2 per 1,000, with the disease being more common in persons of Northern European descent. Idiopathic hemochromatosis is the most common etiology; however, the list of secondary causes is extensive. 90% of cases are due to abnormalities of the HFE gene. Type 2 disease results from mutation of the hemojuvelin or hepcidin genes. Types 3 and 4 disease are caused by mutations of the transferrin receptor gene and ferroportin gene, respectively.
This disease has a strong male predilection, approximately 6:1, although this may be secondary to the fact that women with hemochromatosis do not become symptomatic until later in life because menstruation is therapeutic. In fact female patients with this disease rarely develop symptoms before menopause.
A presumptive diagnosis may be established when there is the characteristic increase in AST and ALT (usually two to five times normal) with an increased transferrin saturation greater than 45-60%(the higher the level the higher the sensitivity) in the presence of a serum ferritin greater than 300 in men and 200 in women. The transferrin saturation can be calculated in the following manner:
Diagnosis maybe confirmed by liver biopsy, which shows micronodular cirrhosis with massive amounts of iron in the parenchymal and Kupffers cells. Cells of the periportal area exhibit maximal iron absorption. The calculated hepatic iron index (hepatic iron [micrograms/grams dry weight] divided by the patients age) may be used to differentiate primary hemochromatosis from iron overload secondary to alcoholic liver disease. A hepatic iron index greater than two is believed to be consistent with hemochromatosis. Plain abdominal films often reveal a right sided double diaphragmatic shadow secondary to excessive hepatic iron stores. An alternative to liver biopsy for confirming the diagnosis is screening for mutations in the HFE gene. Homozygosity for the C282Y (C282Y/C282Y) gene confirms the diagnosis (present in >70% of type 1 cases). H63D homozygotes (H63D/H63D) and compound heterozygotes (C282Y/H63D) are at risk for iron overload but liver biopsy may be required for an accurate diagnosis. Simple heterozygotes rarely have disease. Persons who are younger than 40 years old, are homozygotes for the C282Y gene, a serum ferritin less than 1,000 ng/mL, normal liver testing, and no organomegaly may proceed straight to treatment without biopsy. Patients who do not meet these criteria should have liver biopsy performed prior to treatment.
Treatment of hemochromatosis entails phlebotomy to induce iron deficiency. When treatment is initiated early in the disease course and is continued, a near normal life span may be expected. Phlebotomy is usually initiated weekly until the hematocrit falls to approximately 36% and the ferritin is less than 50. The induction of a mild anemia will stimulate erythropoiesis and therefore mobilization of body iron stores. Patients with chronic transfusion-induced hemochromatosis may not be treated with phlebotomy. Instead continuous subcutaneous infusion of deferoxamine, a chelating agent, with a portable mini-pump results in increased urinary iron excretion. Patients should be offered genetic counseling, and family members should be screened for the disease. Patients should be advised to abstain from eating shellfish because infection and possible sepsis with Vibrio vulnificus is a potential life-threatening complication. Also, phlebotomy therapy does not diminish the probability of hepatocellular carcinoma (HCC); therefore, patients afflicted with this disorder should be screened for HCC via determination of serum alpha-fetoprotein in conjunction with hepatic ultrasonography at regular intervals.
Screening of family members should be instituted. In HFE C282Y homozygotes, family members may be screened with HFE genetic testing along with ferritin levels and transferrin saturation calculation. Complications include cirrhosis and hepatocellular carcinoma.